Pfeiffer syndrom: årsaker, symptomer, behandling
Pfayffer syndrom er ekstremt sjeldenten genetisk sykdom som oppstår i gjennomsnitt i en av 100.000 nyfødte. Like ofte påvirker gutter og jenter. De viktigste symptom på sykdommen - tidlig sammensmelting av kraniet under embryogenesen, på grunn av hvilket hjernen ikke kan utvikle seg normalt i fremtiden.
Generell informasjon
Sykdommen ble oppdaget av den berømte tyskengenetiker Rudolf Pfeiffer. I 1964 beskrev han tidligere ukjente sykdomssymptomer, hovedsymptomene som var uregelmessigheter i hodeskallen struktur og skjøte fingre på føttene eller hendene (syndactyly). Syndrom ble observert i åtte pasienter fra den samme familie, noe som indikerte at det genetisk sykdommens art. Videre studier har vist at Pfeiffer syndrom overføres av en autosomal dominant type.
Ultralyd kan oppdage denne sykdommen i fosteret allerede i andre trimester av svangerskapet. På ultralyd er unormaliteter i utviklingen av skallen, indre organer og ekstremiteter sett.
symptomer
I 1993, den amerikanske genetiker Michael CohenHan foreslo å klassifisere Pfeiffers syndrom i tre typer avhengig av alvorlighetsgraden av symptomene. Nå er klassifiseringen generelt akseptert og mye brukt i medisinsk litteratur.
Pfeiffer syndrom symptomer innbefatter type 1Craniosynostose - en tilstand der en eller flere kraniale suturer fusjonerer for tidlig, danner et holistisk ben. Som et resultat er endringer i form av skallen og unormale ansiktsegenskaper notert - hypertelorisme, lavtliggende ører. Det kan oppstå forringelse av syn, økt intrakranielt trykk, spalt gane. Hos 50% av pasientene er hørselstap registrert. De fleste med denne typen sykdom har et normalt nivå av intelligens og har ikke nevrologiske abnormiteter.
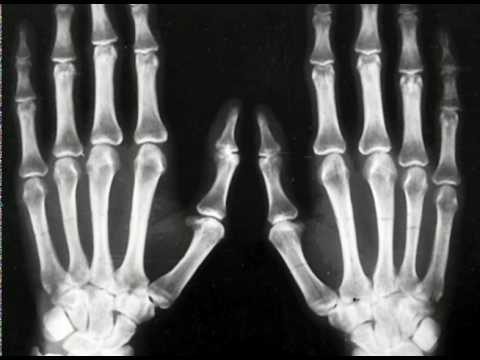
Hos barn med type 1 Pfayffer syndrom, eksternttegn, som korte korte fingre på armer og ben, observeres nesten alltid. Syndaktisk er et mulig, men ikke nødvendig, symptom for denne typen patologi.

Denne typen syndrom er arvelig autosomalt dominant.
Nedenfor er et bilde av Pfeiffer-syndromet. Forventet levetid i Russland for personer som lider av denne sykdommen, overstiger 60 år.

For type 2 sykdom er skallen preget av aform av "trefoil", sammensmelting av ryggvirvlene, syndaktisk og skifting av øyebollene fremover (proptose). Fra de første dagene av livet, ser det alvorlige nevrologiske lidelser opp.
Den tredje typen sykdom karakteriseres enda mertidlig craniosynostosis. Skallen er unormalt langstrakt og ikke danner en "Shamrock". Det er tann anomalier, hypertelorisme, mental retardasjon, misdannelser i de indre organer.
Livstid ved 2. og 3. typesykdommen varierer fra flere dager til flere måneder - defekter av indre organer og nevrologiske lidelser er ikke kompatible med livet. Mutasjonene som er ansvarlige for disse to typer Pfeiffer-syndrom, forekommer sporadisk og er ikke genetisk arvelige.
årsaker
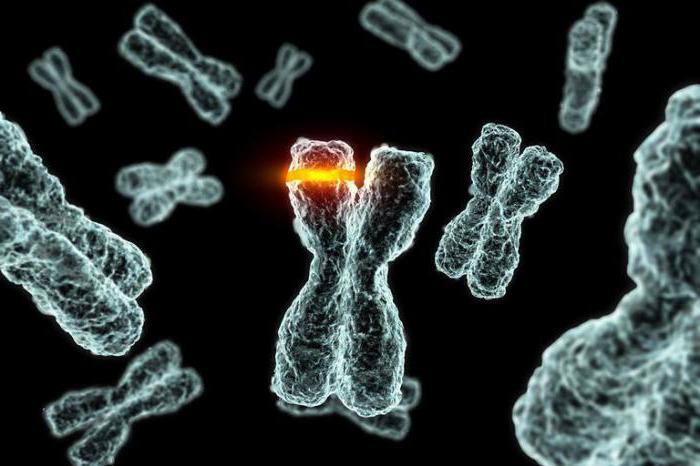
Pfayffer syndrom er assosiert med mutasjoner av reseptor 1fibroblast vekstfaktor (FGFR1) på kromosom 8 eller fibroblast vekstfaktor reseptor 2 (FGFR2) på kromosom 10. De gener som påvirker den mutasjon som er ansvarlig for normal vekst av fibroblaster, som spiller en sentral rolle i kroppens bein utvikling.
behandling
Moderne medisin tillater ikke å elimineremutasjoner i det menneskelige genomet, selv om det er kjent i hvilke gener det har vært perestroika. Derfor, for Pfeiffer syndrom, som for de fleste andre genetiske sykdommer, er behandlingen symptomatisk. For pasienter med type 2 og type 3 er prognosen ugunstig: mange utviklingsmangel kan ikke korrigeres enten medisinsk eller kirurgisk.
Pfayffer type 1 syndrom er kompatibelt med livet. Symptomene på sykdommen - for tidlig fusjon av skallenbenene - kan korrigeres kirurgisk. Operasjonen anbefales for barn til en alder av tre måneder.

I tillegg er kirurgisk inngrep effektiv i tilfelle av simpelt syndaktisk. Behandling er best å begynne i alderen 18-24 måneder, under aktiv vekst av fingre og tær.